생물학적 서열의 기초부터 시퀀싱 기술의 발전, 오류와 호모폴리머 처리까지 시퀀스 데이터의 개념과 취급 방법을 소개한다.
이 논문에서 수행된 작업과 그 과정에서 내려진 선택들을 완전히 이해하기 위해서는 분자생물학과 유전학에 대한 기본 지식이 필요하다. 이미 생물학적 서열에 익숙하다면, 1.2절로 건너뛰어도 좋다.
DeoxyriboNucleic Acid(DNA, 디옥시리보핵산)은 존재하는 분자들 중 가장 중요한 것들 가운데 하나로, 이것 없이는 우리가 아는 복잡한 생명은 불가능하다. DNA에는 특정 생물체의 모든 유전 정보, 즉 그 생물체가 1) 생명체로서 기능하고 2) 자기 자신을 완벽하게 복제하는 데 필요한 모든 정보가 담겨 있다. 이는 지구상의 압도적 다수 생물, 코끼리에서 감자, 박테리아 같은 미생물에 이르기까지 해당된다.
DNA는 중합체로, 뉴클레오타이드라 불리는 단량체 단위들로 구성된다. 각 뉴클레오타이드는 리보스(탄소 5개로 이루어진 당)에 인산기와 네 가지 뉴클레오염기 중 하나가 결합한 형태로 이루어진다. 그 네 가지 염기는 아데닌(A), 시토신(C), 구아닌(G), 티민(T)이다. 이 네 종류의 뉴클레오타이드 단량체는 인산-당 결합을 통해 서로 연결되어 DNA의 단일 가닥을 만든다. 가닥 내에서 이 네 종류 뉴클레오타이드가 정렬된 순서가 생물체가 기능하는 데 필요한 모든 유전 정보를 부호화한다. 한 가닥의 뉴클레오타이드는 다른 가닥의 뉴클레오타이드와 강한 상보적 결합을 형성하는데, A는 T와, C는 G와 결합한다. 이러한 결합 덕분에 두 DNA 가닥이 그림 1.1에 보인 DNA의 이중 나선 구조1를 이룰 수 있다. 뉴클레오타이드 결합의 특이성은 이중 나선의 두 가닥이 서로 상보적임을 보장하며, 한 가닥에 담긴 정보가 다른 가닥으로부터 복원될 수 있음을 보장한다. 이는 DNA 분자에 일정한 구조적 안정성을 제공하고, 가닥 손상으로 인해 중요한 정보가 손실될 수 있는 상황에서 이를 복구하는 방법이 된다.
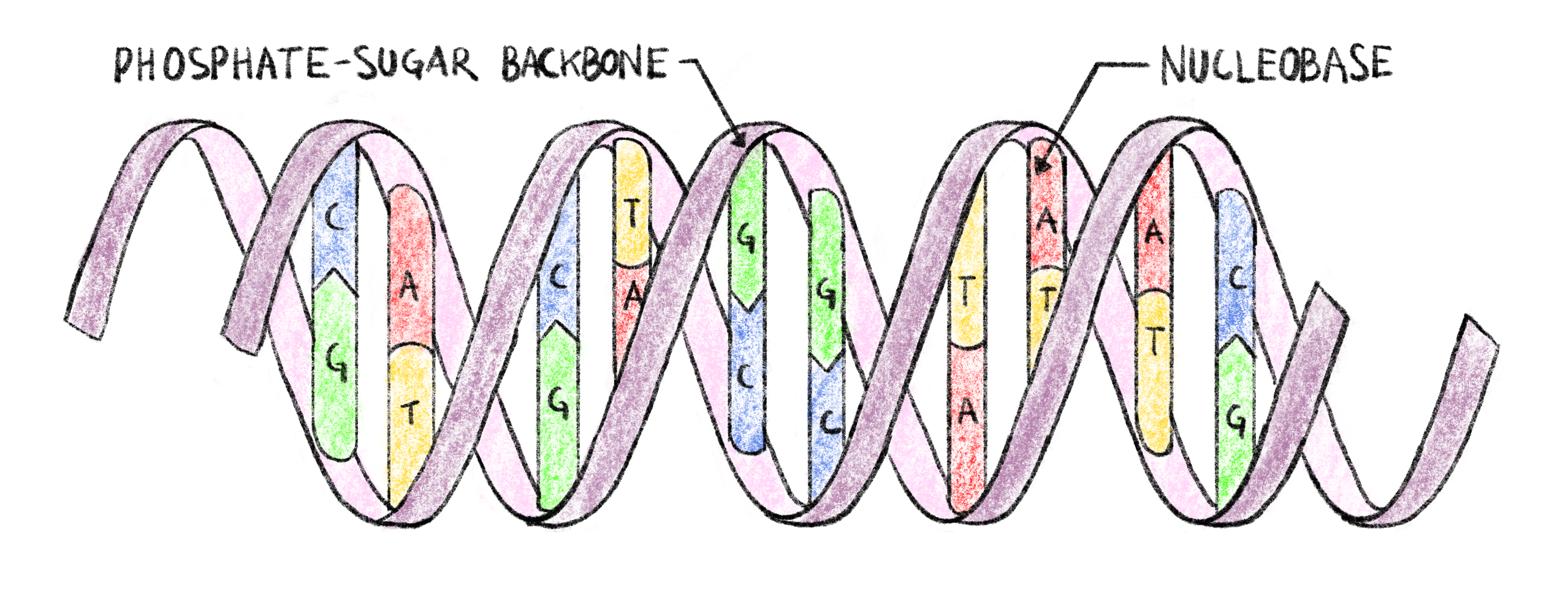
Figure 1.1: DNA의 이중 나선 구조.
각 DNA 가닥은 뉴클레오염기가 부착된 인산-당 골격을 가진다. 두 가닥은 서로 다른 가닥의 뉴클레오염기 사이의 상보적 결합(A는 T와, C는 G와 결합)으로 연결되어 있으며, 두 가닥 모두 동일한 정보를 부호화한다.
정보를 부호화하는 데 필요한 DNA의 양은 생물체마다 크게 다르다. φ X 174 파지2는 5400 염기쌍(5.4kBp), Escherichia coli3는 4.9MBp, Homo sapiens4는 3.1GBp이며, 일본의 산지 개화식물 Paris japonica5는 거의 150GBp에 이른다. 매우 작은 유전체 크기는 대체로 더 작고 단순한 생물에서 나타나는 경향이 있지만, 유전체 크기와 생물 복잡성은 상관관계가 없다6.
생물체 세포 안에 존재하는 이중 가닥 DNA 분자는 정보만을 담고 있다. 생물체가 살아가기 위해서는 이 정보가 읽히고 행동으로 번역되어야 한다. “생명”에 필요한 대부분의 행동은 단백질이라 불리는 큰 분자들에 의해 수행되며, 단백질은 세포 내 반응을 촉매하는 것부터 세포에 구조를 부여하는 것까지 매우 넓은 범위의 기능을 가진다7.
단백질은 하나 또는 여러 개의 아미노산 사슬로 이루어진 거대분자이다. 이 사슬들은 서로 연결되고 특정한 3차원 구조로 접히며, 단백질이 목적을 수행하는 데 필요한 형태를 갖추게 된다. 이 구조는 아미노산 서열에 의해 결정되며, 특정 단백질은 그 아미노산 서열로 식별될 수 있다7.
이 서열은 DNA에 담긴 정보에 직접적으로 의존한다. 먼저 DNA는 유사하지만 단일 가닥인 RNA(Ribonucleic Acid)라는 분자로 전사되며, 이는 동일한 서열을 부호화한다. 이어서 이 RNA 분자는 다음 과정에 의해 단백질로 번역된다8:
뉴클레오타이드가 4종이고 코돈은 3개 뉴클레오타이드를 묶으므로 가능한 코돈은 4 3=64개이다. 그러나 앞서 말했듯 단백질은 20종의 아미노산으로만 이루어져 있어, 서로 다른 여러 코돈이 같은 아미노산에 대응한다. 이는 DNA가 새로운 세포를 만들기 위해 복제될 때, 혹은 단백질 번역에 앞서 RNA로 전환될 때 발생할 수 있는 오류에 대해 번역 과정에 일정한 강건성을 부여한다.
단백질을 만들기 위해 읽히는 DNA의 부분을 “코딩”된다고 하며, 이를 유전자(gene)라고 부른다. 인간 유전체에는 수천 개의 유전자9가 존재하고, 그 결과 세포에서 수천 가지 서로 다른 기능을 수행하는 단백질이 만들어진다. 인간에서는 코딩 DNA가 전체 유전체의 1%~2%에 불과하다10,11. 인간 DNA의 대부분은 단백질로 번역되지 않으며, 그 일부는 전사와 번역을 조절하는 조절 역할을 하지만, 나머지 유전체의 역할은 아직 알려지지 않았다12,13.
DNA 서열에서 단백질로 이어지는 과정은 여러 단계를 포함하는 매우 복잡한 과정이므로 실수가 발생할 수 있다. 유전 정보의 실수와 변형을 피하기 위한 여러 메커니즘이 존재하는데, DNA 두 가닥의 상보성, 유전 부호의 중복성, 그리고 DNA와 RNA를 읽고 쓰는 분자(“polymerases”라 불림)의 오류 수정 메커니즘 등이 그 예이다. 그럼에도 불구하고 핵산(DNA와 RNA)이나 단백질 서열에서 일부 오류는 끝내 걸러지지 않고 남게 되며, 이를 돌연변이(mutations)라고 한다.
유전 정보를 변화시킬 수 있는 오류의 원인은 여러 가지가 있다14:
DNA 복제: 세포가 분열하거나 생물이 번식할 때, 유전 정보를 보존하고 전달하기 위해 DNA 분자를 복제해야 한다. 이 과정의 오류율은 매우 낮아, 복제된 염기쌍 10억 개에서 1000억 개당 오류 1개 수준까지도 보고된다15. 이는 DNA 분자를 복사하는 단백질인 DNA 폴리머레이스 자체의 오류율이 비교적 낮기 때문이기도 하지만, 무엇보다 특정 세포와 박테리아에 존재하는 오류 수정 메커니즘 덕분이다16.
RNA 전사: RNA 전사체의 오류는 복제된 DNA에서의 오류보다 중요성이 낮기 때문에, RNA 폴리머레이스의 오류율은 DNA 폴리머레이스보다 훨씬 높다. 이 오류율은 전사된 염기 100만 개당 4개17에서 10만 개당 2개18 사이로 추정된다.
단백질 번역: RNA를 단백질로 번역하는 과정은 리보솜이라 불리는 단백질에 의해 수행된다. 이는 오류가 많이 발생하는 과정으로, 번역된 코돈 10,000개당 1개 수준의 오역(mistranslation)률로 추정된다19
기타 돌연변이 유발 사건: 이온화 방사선20, 자외선21, 독소22, 열 스트레스23, 냉 스트레스24, 산화 스트레스25 등 많은 외부 사건과 요인이 노출된 DNA에서 돌연변이를 유발하는 것으로 알려져 있다.
RNA 전사와 단백질 번역은 DNA 복제보다 훨씬 오류가 많이 발생하지만, 이들 오류는 유전 정보의 발현만 변화시킨다. 이러한 오류의 영향은 발생한 세포에 국한되며 자손에게 전달되지 않는다. 그러나 전사 오류가 중요하지 않다는 뜻은 아니며, 전사 오류율 증가가 소아 코호트에서 심각한 신경학적 증상을 유발할 수 있다는 가설도 제기되었다26.
생물학적 서열(핵산과 단백질)에서 돌연변이는 다음 세 가지 오류 양상 중 하나로부터 발생할 수 있다:
이 세 가지 오류 유형은 핵산과 단백질 모두에서 발생하지만, 핵산 돌연변이가 단백질 합성에 미치는 결과에 대해서는 몇 가지 고려할 점이 있다. 1.1.2.1절에서 언급한 유전 부호의 중복성 때문에, 핵산 서열의 일부 치환은 동일한 단백질 서열을 만들어 단백질 활성에 변화를 주지 않을 수 있다. 그러나 일부 돌연변이는 아미노산 수준의 치환으로 이어져 물리화학적으로 변형되거나 기능을 상실한 단백질을 만들 수 있다. 마지막으로 삽입과 결실 오류(통칭 indels)는 생성되는 단백질에 큰 영향을 미칠 수 있다. 뉴클레오타이드를 3의 배수로 삽입하거나 삭제하면 단백질에서 아미노산의 삽입/삭제로 이어지지만, 그 외 길이의 indel은 프레임시프트 돌연변이(frameshift mutation)27를 일으킨다. 이러한 돌연변이는 돌연변이 이후의 모든 코돈을 변화시키며, 그림 1.2에서 보듯 조기 종결 코돈의 출현을 포함해 완전히 다른 아미노산 서열을 초래할 수 있다.
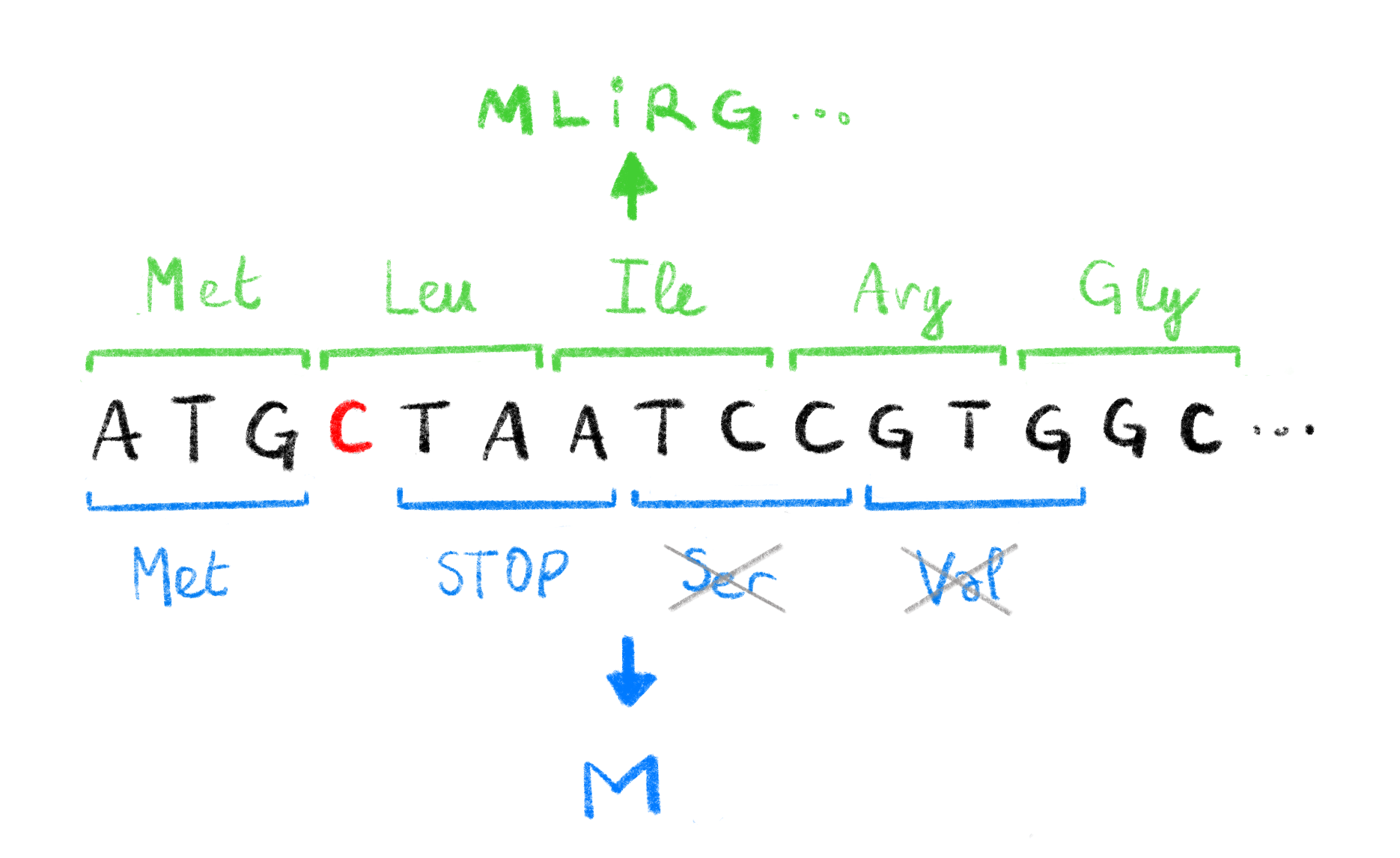
Figure 1.2: 프레임시프트 돌연변이의 영향.
원래 DNA 서열에서 단일 C(빨간색으로 강조됨)를 삭제하면 번역 중 읽히는 코돈이 변한다. 원래 코돈(초록색으로 표시되고, 대응 아미노산이 서열 위에 표시됨)은 기능성 단백질 MLIRG...로 번역된다. 삭제로 인해 새로 생긴 코돈(파란색으로 표시되고, 대응 아미노산이 서열 아래에 표시됨)은 조기 STOP 코돈을 유도하여 비기능성 단백질 M을 만든다. 세린과 발린 코돈은 STOP 코돈 때문에 번역되지 않는다.
앞서 말했듯 DNA의 일부 돌연변이는 아무 영향이 없을 수 있지만, 다른 일부는 비기능성 단백질을 초래할 수 있다. 어떤 경우에는 돌연변이가 변이 개체의 형질과 연관될 수 있다. 예를 들어 응고와 관련된 유전자의 단일 돌연변이가 병적 Leiden thrombophilia28를 유발할 수 있고, CFTR 단백질에서 단일 아미노산 결실은 (매우 치명적인) 낭성 섬유증29을 유발한다. 또한 많은 돌연변이가 제2형 당뇨병 같은 복합 질환과 연결되어 왔다30,31. 하지만 모든 돌연변이의 효과가 생물체에 나쁜 것은 아니며, 돌연변이는 박테리아32나 HIV 같은 바이러스33가 치료에 대한 내성을 획득하는 데 필수적이다(이에 대해서는 5장과 6장에서 더 다룬다).
일부 돌연변이는 그 메커니즘과 결과가 철저히 연구되었지만, 많은 경우 돌연변이는 단지 어떤 형질과 연결되어 있을 뿐이다. 상관관계를 보이는 것이 인과관계를 보이는 것보다 쉽고, 전자가 반드시 후자를 함의하지는 않기 때문에, 주목할 만한 돌연변이에 대해서는 잠재적 결과를 이해하기 위해 추가 연구가 중요하다.
많은 분야, 특히 계산생물학에서 우리는 연구 대상 생물체가 어떤 유전 정보를 갖는지 알아야 한다. 즉, 그 생물체의 DNA를 구성하는 뉴클레오타이드의 정확한 서열이 무엇인지 알아야 한다. 이 서열을 알아내는 과정은, 놀랍지 않게도, 시퀀싱(sequencing)이라 불린다. 이 과정으로 생산된 서열은 sequencing read 혹은 더 흔히 _read_라고 부른다.
최초로 널리 사용된 시퀀싱 방법은 1977년에 개발되었다34. Sanger _et al._은 DNA 서열을 구성하는 뉴클레오타이드의 순서를 읽는 간단한 방법을 고안했는데, 이는 chain termination sequencing 또는 간단히 _Sanger sequencing_이라 불린다(그림 1.3 참조). 이 방법은 현재 대부분 구식이지만, 시퀀싱의 핵심 개념들을 확립했으며 그 중 일부는 현대의 시퀀서에서도 여전히 사용된다.
Sanger 시퀀싱을 이해하려면 먼저 DNA를 합성하는 방법을 이해해야 한다. 1.1.1절에서 말했듯 DNA는 뉴클레오타이드, 더 구체적으로는 디옥시뉴클레오타이드 삼인산(deoxynucleotide triphosphates, dNTPs)이라 부르는 구성 블록으로 쌓인다. dNTP는 당(디옥시리보스), 뉴클레오염기(A, T, G 또는 C), 그리고 인산기 3개로 이루어진다. 기존 DNA 분자의 끝에 이러한 dNTP를 연속적으로 추가하면 DNA가 연장되는데, dNTP의 인산기 중 하나가 DNA 분자 마지막 뉴클레오타이드의 산소 원자와 연결된다. 이제 디디옥시뉴클레오타이드 삼인산(dideoxynucleotide triphosphate, ddNTP)을 생각해보자. ddNTP는 특정 산소 원자 하나가 제거된 것을 제외하면 dNTP와 동일하다. ddNTP도 일반 dNTP처럼 성장하는 DNA 분자에 추가될 수 있지만, 그 산소 원자가 없기 때문에 이 뉴클레오타이드 이후에는 더 이상의 dNTP나 ddNTP가 DNA에 붙을 수 없다. 연장이 종료되며, 이러한 ddNTP를 체인-터미네이터(chain-terminators)라고 부른다. DNA 합성과 그 뒤의 종결의 결합이 Sanger 시퀀싱의 핵심이다.
dNTP와 ddNTP는 어떤 뉴클레오염기를 갖든 모두를 가리키는 점이 중요하다. 특정 dNTP를 말할 때는 “N”을 원하는 염기로 바꿔 표기한다. 예를 들어 dATP는 아데닌을 염기로 갖는 dNTP를 뜻한다. 마찬가지로 dCTP, dGTP, dTTP(그리고 ddATP, ddCTP, ddGTP, ddTTP)도 있다.
이 과정은 Sanger _et al._이 1977년에 φ X 174 박테리오파지의 최초 유전체를 시퀀싱하는 것을 가능하게 했다2. 혁명적이긴 했지만, 이 방법은 비용이 많이 들고 시간이 오래 걸리며 노동집약적이었다. 이를 더 빠르고 저렴하게 만들기 위해 여러 조정이 이루어졌다. 중요한 단계 중 하나는 ddNTP의 표지 방식을 바꾸는 것이었다. 각 염기마다 서로 다른 “색”을 갖는 형광 표지를 사용하면, 겔에서 4개의 서로 다른 환경과 레인이 필요 없어진다35,36. 이는 또한 시퀀싱 자동화의 길을 열었는데, 형광 표지된 각 밴드는 레이저로 여기될 수 있고, 그 결과 특정 파장이 광학 시스템에 의해 기록되며, 대응 염기가 자동으로 추론된다37(그림 1.3도 참조). 그 밖에도 겔 전기영동 대신 모세관 전기영동을 사용하는 등의 개선이 있었다.
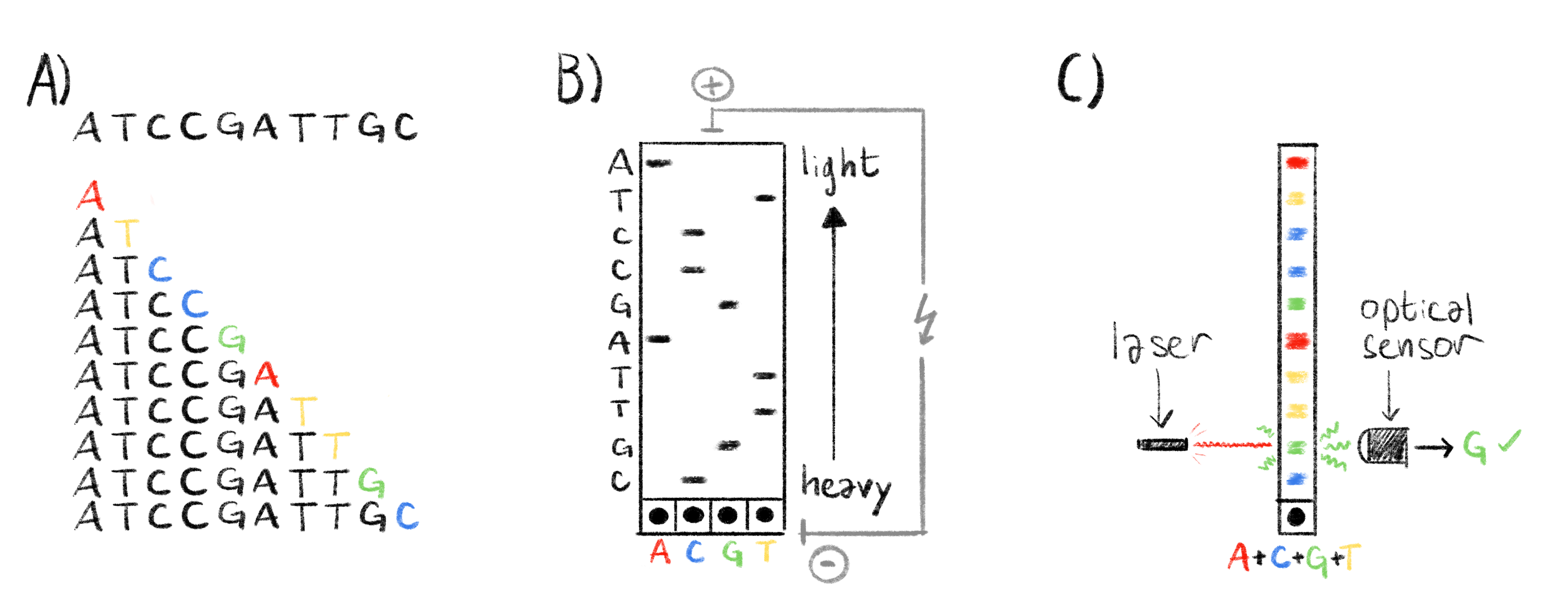
Figure 1.3: Sanger 시퀀싱 프로토콜 개요.
A) 읽어야 하는 서열과 생성된 모든 조각들. ddNTP 체인 터미네이터가 강조되어 있으며 분자량(즉 길이) 순으로 정렬되어 있다. B) 고전적 Sanger 시퀀싱. 조각들은 전기영동으로 분리되며 더 가벼운 조각이 겔 하단의 웰에서 더 멀리 이동한다. 겔의 각 레인은 특정 ddNTP에 대응한다. 방사성 표지된 ddNTP는 겔에서 검은 밴드로 나타나며, 위에서 아래로 밴드를 읽고 밴드가 나타나는 열이 각 위치의 염기를 나타내므로 서열을 재구성할 수 있다. C) 자동화 Sanger 시퀀싱. 조각들은 B 패널과 마찬가지로 전기영동으로 분리된다. 체인 터미네이터는 형광 표지로 표시된다. 레이저로 여기되면 각 ddNTP는 특정 파장을 방출한다. 이는 광학 센서가 읽고 대응 ddNTP가 기록된다. 각 밴드를 여기함으로써 빠르게 서열을 유도할 수 있다.
Sanger 시퀀싱 프로토콜의 이러한 점진적 개선은 더 길고 더 정확한 read를 시퀀싱하는 것을 가능하게 했으며, 최신 기술에서는 1,000 염기쌍에 달하는 read를 99.999%의 정확도로 얻을 수 있다38. 또한 이러한 개선은 시퀀싱 비용을 크게 낮추어, 염기쌍당 약 $100039에서 킬로베이스당 $0.538로 줄였다. 마지막으로 이러한 기술 발전은 시퀀싱 기기의 처리량을 하루 약 1킬로베이스39에서 시간당 120킬로베이스40까지 끌어올렸다.
이러한 개선에도 불구하고 인간 유전체 프로젝트 같은 야심 찬 사업에서 시퀀싱은 거대한 과업이었다. 최초 인간 유전체를 시퀀싱하는 데 든 비용은 5억~10억 달러로 추정된다41.
이러한 대규모 시퀀싱 프로젝트와 유전체학 분야 전반 덕분에 서열 데이터의 풍부함과 유용성이 점점 더 분명해졌다. 서열 데이터에 대한 수요 증가는 Sanger 시퀀싱보다 더 높은 처리량과 더 낮은 비용을 목표로 한 새로운 시퀀싱 방법들의 개발로 이어졌고, 이는 시퀀싱의 새로운 시대를 열었다. 이러한 2세대 시퀀싱 기술을 흔히 Next-Generation Sequencing(NGS) 또는 Massively parallel 시퀀싱이라고 부른다. 기술은 다양하지만 몇 가지 공통된 핵심 요소를 공유한다42:
Sanger 시퀀싱과 마찬가지로 DNA 템플릿을 먼저 증폭하고 클로닝해야 한다. 다만 이들 기술은 Sanger 시퀀싱보다 더 짧은 read를 생성하므로, 시퀀싱할 DNA를 먼저 무작위로 작은 템플릿 조각으로 분절한 뒤 증폭해야 한다.
증폭된 템플릿 조각들은 어떤 형태의 고체 지지체에 부착되어, 수십억 개의 템플릿 조각이 붙어 있는 물리적 지지체가 만들어진다.
Sanger 시퀀싱과 마찬가지로 템플릿 조각과 상보적인 DNA 분자가 연장된다. 이는 수십억 개의 조각에 대해 동시에 일어난다(따라서 “massively parallel”이라는 이름이 붙었다).
사슬에 특정 뉴클레오타이드가 추가되는 것을 실시간으로 검출하며, 영구적인 체인 종결은 없다. 전기영동이라는 긴 단계가 필요하지 않다. 검출은 동시에 연장되는 모든 분자에 대해 동시에 일어난다.
이 단계들의 결과는 매우 많은 수의 짧은 read이다. 데이터 분석을 통해 이 짧은 read들은 더 긴 서열을 유추하는 데 사용될 수 있으며, 결국 _assembly_라 불리는 과정을 통해 원래 전체 유전체 서열의 분절된 근사치를 구성할 수 있다.
주요 NGS 방법은 Illumina라는 회사가 개발한 “sequencing by synthesis”이며, 일반적으로 _Illumina sequencing_이라 불린다. 이 방법은 90년대 Institut Pasteur에서 개발된43 _reversible chain terminators_에 기반한다. 이는 DNA 분자를 연장하는 데 사용할 수 있는 표지된 dNTP이지만, 추가적인 분자기(molecular group)를 가지고 있어 기본적으로는 터미네이터로 작동한다. 그러나 이 종결 그룹은 NTP가 DNA 분자에 포함된 뒤 제거될 수 있어, 연장 과정이 계속될 수 있다. 이 dNTP들은 형광 표지되어 있으며 레이저로 여기되면 고유한 색의 빛을 방출한다. Illumina 시퀀싱에서는 이 가역적 체인 터미네이터가 수백만 개 조각에 동시에 포함되어 연장을 멈춘다. 그 시점에서 모든 조각을 레이저로 여기하고, 광학 시스템이 동시에 모든 조각의 방출 색을 사진으로 기록한다. 이 이미지에서 픽셀은 대략 시퀀싱된 조각에 대응하고, 그 색은 가장 최근에 추가된 dNTP에 대응한다. 이후 종결 그룹을 절단하고, 새로운 배치의 가역적 터미네이터를 포함시키며 과정을 반복한다. 연속된 이미지들을 관찰하면 각 조각에 대해 추가된 뉴클레오타이드의 서열을 유추하여 read들을 얻을 수 있다.
또 다른 NGS 방법으로 454 Life Sciences가 상용화한 pyrosequencing이 있다. Illumina 시퀀싱과 달리 이 방법은 가역적 체인 터미네이터를 사용하지 않는다. 대신 특정 dNTP가 추가될 때 빛을 방출하는 luciferase라는 특수 효소를 사용한다. 이 과정은 4종 dNTP에 대해 반복되며(Sanger 시퀀싱과 유사하게), 빛 방출로부터 뉴클레오타이드 서열을 유추할 수 있다44.
이러한 기술들은 Illumina의 경우 약 150 뉴클레오타이드, pyrosequencing의 경우 400nt 정도의 read를 산출한다45. 이는 최신 Sanger 시퀀싱 기술로 얻을 수 있는 1kB read보다 훨씬 짧다. 그러나 처리량은 훨씬 높다40: Illumina는 시간당 2.5~12.5 기가베이스, pyrosequencing은 시간당 30 메가베이스. 비용도 낮아 Illumina는 메가베이스당 $0.07, pyrosequencing은 메가베이스당 $10 수준이다. 염기 단위 시퀀싱 정확도 역시 매우 높아, Illumina46와 pyrosequencing40 모두 최대 99.9%까지 보고된다. 다양한 시퀀싱 기술의 핵심 특성 요약은 표 1.1에 제시되어 있다. 더 낮은 비용과 더 높은 처리량으로 인해 Illumina 시퀀싱 기술이 지배적인 기술이 되었으며, 회사는 2015년에 전 세계 시퀀싱 데이터의 90%가 Illumina 장비로 생성되었다고 추정한다47.
NGS 기술이 시퀀싱 세계를 혁신했지만, 최근에는 더 긴 read를 얻기 위한 노력이 이어지고 있다. 이러한 3세대 방법은 수십 킬로베이스 길이의 read를 생성하며, 흔히 long-read sequencing 방법이라고 부른다. 롱리드는 다양한 응용을 가진다48. 짧은 NGS read가 적합하지 않을 수 있는 응용들로는 대형 복잡 유전체의 De novo 어셈블리, 동원체(centromeres)나 텔로미어(telomeres) 같은 복잡 반복 영역 연구, 구조 변이(structural variants) 탐지 등이 있다. 최근에는 텔로미어 및 동원체 영역을 포함해 최초의 진정한 완전 인간 유전체를 조립하는 데 사용되기도 했다4.
현재 이용 가능한 롱리드 기술은 Pacific Biosciences(PacBio)가 상용화한 Single Molecule Real Time sequencing(SMRT)과 Oxford Nanopore Technologies(ONT)가 상용화한 Nanopore 시퀀싱이다. 두 기술은 꽤 다르지만, 둘 다 체인 터미네이터나 별도의 시퀀싱 반응 없이 실시간으로, 높은 처리량과 비교적 낮은 비용으로, Sanger 시퀀싱보다 훨씬 긴 read를 산출한다.
SMRT 시퀀싱은 2009년에 처음 개발되었고49, 이후 PacBio가 상용화하며 발전시켰다. 기본 원리는 다음과 같다:
Nanopore 시퀀싱은 1980년대에 구상되어 수년간 발전했으며51, 2014년에 ONT에 의해 처음 상용화되었다52. 이는 앞서 언급한 모든 시퀀싱 기술과 완전히 다르다. 다른 기술들이 모두 상보적 DNA 가닥을 합성하고 특정 dNTP의 포함을 어떤 방식으로든 검출하는 데 기반한 반면, nanopore 시퀀싱에는 합성이 없다. 원리는 단일 가닥 DNA 템플릿을 막(membrane)에 있는 작은 구멍, 즉 _nanopore_를 통해 제어된 속도로 통과시키는 것이다. 뉴클레오타이드가 nanopore를 통과할 때 막의 양쪽 사이에 전류가 형성된다. 이 전류는 측정될 수 있으며, 특정 시점에 nanopore 채널 내부에 있는 5~6개 뉴클레오타이드의 연속에 특이적이다. DNA 가닥이 nanopore를 통과하는 동안 전류의 변화를 보면, _base calling_이라 불리는 과정을 통해 뉴클레오타이드 서열을 유추할 수 있다. Base calling은 보통 기계학습 방법, 주로 인공 신경망에 의해 수행된다53. ONT 시퀀서에서 사용되는 플로우셀(flow cells)에는 합성 막 위에 수십만 개의 nanopore가 분포해 있어, 이 역시 massively parallel 시퀀싱이 가능하다. 이론적으로 이 방법은 합성에 기반하지 않으므로 read 길이의 상한은 템플릿의 길이에 의해서만 제한되며, 실제로 ONT 시퀀싱은 가장 긴 read를 생성한다.
두 기술 모두 롱리드를 생성한다. PacBio 시퀀싱의 경우 read 길이의 중앙값과 최대값은 각각 10킬로베이스와 60킬로베이스로 보고된다54. Nanopore의 중앙 read 길이 10~12킬로베이스55,56는 PacBio와 유사하지만, 1에서 2.3 메가베이스 길이의 초장기(ultra-long) read도 산출할 수 있다57–59. read 길이와 병렬성 덕분에 두 시퀀서의 처리량은 매우 크다. PacBio 시퀀서는 시간당 2~11 기가베이스를, ONT는 시간당 12.5 기가베이스에서 최신 ONT PromethION 장비로는 무려 시간당 260 기가베이스까지 시퀀싱할 수 있다56. Illumina 시퀀서보다 비용이 높기는 하지만, 이들 장비의 시퀀싱 비용은 PacBio와 ONT가 각각 메가베이스당 $0.32와 $0.13으로 여전히 합리적인 수준이다60. 이러한 특성은 다른 시퀀싱 기술과 함께 표 1.1에 요약되어 있다.
이 두 기술의 길이, 처리량, 비용은 매우 매력적으로 보이며 실제로 많은 환경에서 유용함이 입증되었다. 그러나 시퀀싱 정확도는 이들 기술의 문제이다. 염기 단위 시퀀싱 정확도는 PacBio 시퀀서가 85%~92%, ONT 장비가 87%~98%로 추정된다56,61,62. 이는 Sanger 시퀀싱이나 Illumina read보다 훨씬 낮다. 이러한 오류를 특성화하고, 교정하며, 분석에서 고려하는 방법은 널리 연구되고 있으며, 1.3절과 1.4절에서 더 자세히 논의한다.
Table 1.1: 시퀀싱 기술 특성 비교.
Sanger 시퀀싱 항목에는 최신 시퀀서의 특성을 사용했다. 길이는 뉴클레오타이드 단위, 처리량은 시간당 시퀀싱된 뉴클레오타이드 수, 비용은 메가베이스당 미국 달러로 표시했다.
언급된 대부분의 기술은 DNA 대신 RNA를 시퀀싱하도록도 적용할 수 있지만63,64, 단백질을 직접 시퀀싱하는 것은 여전히 도전 과제다. 단백질을 구성하는 아미노산 서열은 보통 시퀀싱된 DNA 또는 RNA의 코돈에서, open reading frames(ORFs)라 불리는 잠재적 코딩 영역을 탐지한 뒤 유추된다. 질량분석을 이용해 단백질 분자를 직접 시퀀싱하는 방법의 개발은 Sanger 시퀀싱 이후 그리 오래 지나지 않아 시작되었고65 개선되었다66. 새로운 방법도 여전히 개발되고 있지만67, 단백질 시퀀싱은 여전히 어려운 과제다.
시퀀싱 기술은 완벽하지 않다. 1.2절에서 보고된 정확도에서 보듯 오류가 발생한다. 핵산 합성에 기반한 기술(즉 ONT를 제외한 모든 기술)의 경우 폴리머레이스를 사용하므로, 1.1.2.2절에서 설명한 동일한 세 가지 오류 유형(치환, 삽입, 결실)이 발생하는 것이 합리적이다. 그러나 롱리드 기술의 경우 오류의 대부분은 폴리머레이스가 아니라, 서열을 유추하기 위해 사용되는 신호 처리에서 비롯된다. 두 기술 모두 단일 분자 시퀀싱을 수행하기 때문에 신호 대 잡음비가 낮아68,69 base calling이 더 복잡해진다.
이는 짧은 read와 긴 read 시퀀싱 기술 사이의 오류율 차이를 설명한다. 전자는 계산적 처리 이후 10-4 또는 10-5까지 낮아질 수 있는 반면70, 후자는 10%~15% 수준이다. 이러한 높은 오류율의 롱리드는 부담스럽고, 이를 낮추기 위한 많은 노력이 계산적·기술적으로 이루어졌다.
롱리드 오류 교정에 관한 문헌과 도구 생태계는 풍부하고 활발하다71–73. 오류를 교정하는 주요 방법은 두 가지가 있다: 1) 높은 정확도의 짧은 read를 사용하는 하이브리드 방법, 2) 롱리드만 사용하는 비하이브리드 방법.
비하이브리드 방법71,74에서는 read들 사이에 상당히 잘 겹치는 영역을 찾고, 겹친 영역의 컨센서스(즉 각 위치에서 다수의 뉴클레오타이드)를 취함으로써 일부 오류를 제거할 수 있다. 많은 분석 및 시퀀싱 데이터 처리 파이프라인에서 첫 단계는 read를 k 길이의 가능한 모든 겹치는 부분서열인 k-mer로 분해하는 것이다(예: ATTGC의 3-mer는 ATT, TTG, TGC). read 데이터셋에서 드물게 나타나는 k-mer, 즉 전체 read에서 몇 번만 등장하는 k-mer는 오류의 결과일 가능성이 높으며, 이를 필터링하면 분석이 개선될 수 있다. 이러한 절차 중 하나 또는 둘 다는 wtdbg275, canu76 같은 어셈블러나 daccord77 같은 독립형 롱리드 교정기 등 여러 널리 쓰이는 소프트웨어에 구현되어 있다. 어떤 경우에는 원시 read가 아니라 롱리드를 긴 연속 서열(contigs)로 어셈블한 뒤에 오류를 교정하는데, 이를 polishing이라 한다. ntEdit polisher78도 드문 k-mer를 필터링하여 오류를 교정한다. Arrow79와 Nanopolish80 polishers는 각각 원시 PacBio 및 ONT 롱리드를 사용해 어셈블리를 교정하며, Racon81은 두 종류의 롱리드 모두를 사용해 어셈블리를 polishing할 수 있다.
하이브리드 방법은 이름 그대로 짧은 read를 이용해 롱리드의 오류를 교정한다. 짧은 read와 긴 read 사이의 유사 영역을 찾음으로써, 더 정확한 짧은 read를 사용해 긴 read를 교정할 수 있다. 이는 proovread82, Jabba83, PBcR84, LoRDEC85 등 많은 소프트웨어에 구현되어 있다. 짧은 read는 Pilon86 같은 도구로 롱리드 어셈블리를 polishing하는 데에도 사용될 수 있다. 최초의 완전 인간 유전체는 PacBio, ONT, Illumina를 포함한 여러 시퀀싱 기술을 사용해 조립 및 polishing되었다4.
오류 교정에 많은 노력이 투입되는 한편, 롱리드 오류율을 낮추기 위한 또 다른 접근은 시퀀싱 기술 자체를 개선하는 것이다.
2019년 PacBio는 원형 컨센서스(CCS) 기법87에 기반한 HiFi read를 도입했다. SMRT 시퀀싱에서는 두 가닥이 bell adapter로 연결되어 원형 DNA 템플릿을 형성하는데(1.2.3절 참조), CCS의 핵심은 이 분자를 원을 한 번 이상 돌며 여러 차례 시퀀싱하는 것이다. 결과로 얻은 긴 서열에서 알려진 bell adapter 서열은 제거할 수 있고, 동일 템플릿을 여러 번 통과한 결과로부터 컨센서스 서열을 구축할 수 있다. 이는 99.8%~99.9%의 롱리드 정확도를 제공한다56,87. 이는 PacBio 시퀀싱 오류가 주로 시퀀싱된 템플릿을 따라 무작위로 분포하기 때문에 가능하다(1.4.2절에서 더 다룸). 따라서 동일한 오류가 같은 템플릿 구간에서 여러 패스에 걸쳐 반복될 가능성은 낮다.
ONT 시퀀싱의 경우, 개선 노력의 대부분은 base-caller에 집중되어 왔다. 이 도구들은 원래 Hidden Markov Models(HMMs)88에 기반했으나, 점차 더 빠른 추론 시간과 더 나은 성능을 가진 신경망 기반 딥러닝 방법53,74,89,90으로 이동하고 있다.
PacBio HiFi read와 유사하게, ONT는 2D 및 1D 2 시퀀싱을 개발했다. 2D 시퀀싱에서는 시퀀싱할 DNA 분자의 양 가닥을 헤어핀 어댑터(hairpin adapter)로 연결해 하나의 긴 서열을 만든다. 각 가닥을 한 번씩 시퀀싱하고, 이 2회 패스로부터 컨센서스를 구축한다91. 1D 2 시퀀싱은 헤어핀 어댑터 없이 유사하게 동작한다92. 2D 시퀀싱은 표준 1D 시퀀싱보다 훨씬 짧지만 97% 정확도의 read를 생성한다91. 최근 Oxford Nanopore Technologies는 duplex라는 새로운 기술의 출시를 발표했다. 새로운 화학, 새로운 basecaller, 그리고 양 가닥 시퀀싱(2D 및 1D 2와 유사)을 사용해 원시 read 정확도 99.3%를 발표했다93. 사전 인쇄된 연구는 이러한 수치를 대체로 확인하는 것으로 보이며, 한 실험에서는 duplex read 정확도 99.9%가 보고되었다94.
템플릿 준비 단계에서 unique molecular identifiers를 추가하고 컨센서스 시퀀싱을 수행하는 기술 중립적 방법은, 특정 맥락에서 ONT 및 PacBio CCS 롱리드의 정확도를 각각 99.59%와 99.93%로 향상시킬 수 있음이 보였다95.
마지막으로, 오류 보정이 내장된(short-read) 기술로 최대 200 뉴클레오타이드 길이의 무오류 read를 산출하는 새로운 시퀀싱 기술도 개발 중이다96. Illumina 또한 2022년에 자체적인 고처리량·고정확도 롱리드 시퀀싱 기술을 발표했지만97, 성능과 기술의 상세 정보는 많지 않다.
오류 교정 방법과 시퀀싱 기술이 개선되고 있음에도 불구하고, 특정 유전 패턴은 처리하기 특히 어렵다. 호모폴리머는 그러한 패턴 중 하나이다.
_호모폴리머(homopolymers)_는 유전체의 어떤 지점에서 연속으로 반복되는 뉴클레오타이드(즉 ≥2)로 이루어진 구간이다. 예를 들어 AAAA 서열은 길이 4의 아데닌 호모폴리머이다. 완전 인간 유전체 어셈블리(T2T 컨소시엄의 CHM13 v1.14)에서는 30억 염기의 50%가 길이 2 이상의 호모폴리머에 속하며, 10%는 길이 4 이상의 호모폴리머에 속한다. 그림 1.4에서 보듯, 짧거나 중간 길이의 호모폴리머는 유전체의 상당 부분을 차지한다. 이전의 GRCh38 인간 유전체 어셈블리에서는 길이 8 이상 호모폴리머에 속하는 부분이 1.9 메가베이스를 넘으며98, 이는 그 어셈블리의 약 1‰에 해당한다. CHM13 v1.1 어셈블리에서 가장 긴 호모폴리머 런(run)은 86이다(GRCh38에서는 9098).
![Image 4: 호모폴리머 길이에 따른 전체 인간 유전체의 호모폴리머 비율.호모폴리머 카운트는 T2T 컨소시엄의 완전 인간 유전체 어셈블리 CHM13 v1.1에서 계산되었다. 이 그림은 참고문헌 [@booeshaghiPseudoalignmentFacilitatesAssignment2022]의 Figure 3b에서 영감을 받았다.](https://thesis.lucblassel.com/_main_files/figure-html/HPpercent-1.png)
Figure 1.4: 호모폴리머 길이에 따른 전체 인간 유전체의 호모폴리머 비율.
호모폴리머 카운트는 T2T 컨소시엄의 완전 인간 유전체 어셈블리 CHM13 v1.1에서 계산되었다. 이 그림은 참고문헌98의 Figure 3b에서 영감을 받았다.
인간 유전체에서 호모폴리머는 구아닌과 시토신의 연속보다 아데닌과 티민의 연속에서 더 자주 나타나는 경향이 있다. A 또는 T 호모폴리머(481 Mb와 484 Mb) 안에 포함된 뉴클레오타이드는 G 또는 C(278 Mb와 279 Mb)보다 약 두 배 많다. 이 차이는 길이 4보다 긴 호모폴리머만 보면 더욱 두드러진다(그림 1.5 참조).
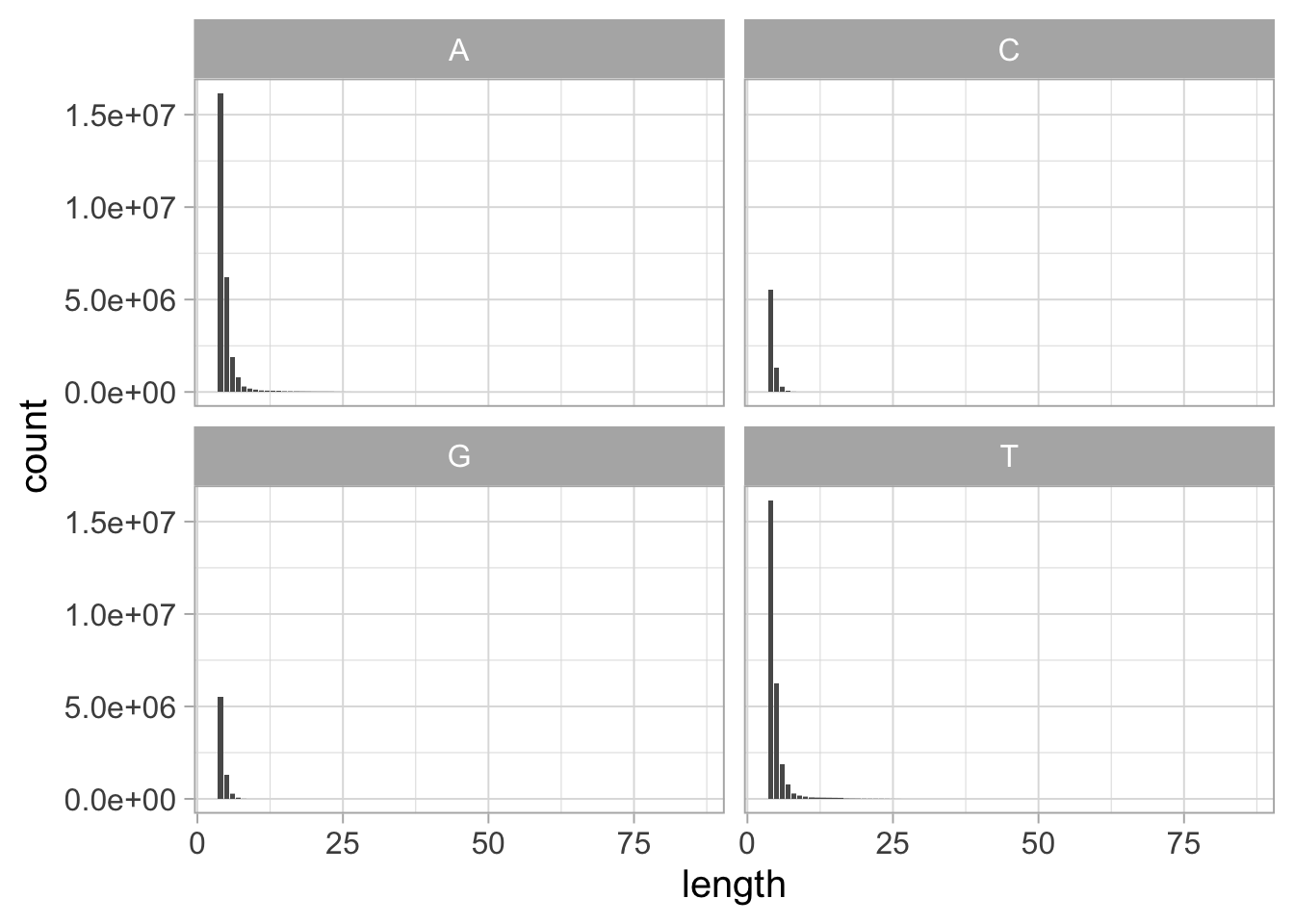
Figure 1.5: 인간 유전체에서 염기별 호모폴리머 길이 분포(길이 ≥ 4인 호모폴리머).
호모폴리머 카운트는 T2T 컨소시엄의 완전 인간 유전체 어셈블리 CHM13 v1.1에서 계산되었다.
불행히도 호모폴리머는 시퀀싱 오류의 원천이며, 특히 롱리드 기술에서 그렇다. PacBio와 ONT에서 치환 오류는 read를 따라 무작위로 분포하는 것으로 보이지만, 주요 오류 양상은 호모폴리머 구간에서의 indels, 즉 같은 뉴클레오타이드를 여러 번 읽거나 반복된 뉴클레오타이드 중 하나를 건너뛰는 것으로 보인다. 많은 연구에서 호모폴리머 indel이 PacBIO SMRT 및 ONT 롱리드 시퀀싱의 주요 오류 유형임을 보여준다68,99–101. 이는 PacBio HiFi read에서도 마찬가지인데, 원형 컨센서스 접근은 무작위로 분포한 치환을 제거하지만 호모폴리머 indel은 남는다87. ONT read가 PacBIo보다 이 오류 유형에 더 취약한 것으로 보인다56. ONT에서는 이러한 오류율이 호모폴리머 길이와 무관하지만, 짧은 read 및 PacBio 기술에서는 호모폴리머 길이가 길어질수록 오류율이 증가한다102.
호모폴리머가 인간 유전체의 상당 부분을 차지하고, 롱리드 기술에서 오류의 원천이 된다는 사실은 호모폴리머에 대해 특별한 주의와 처리가 필요함을 의미한다. 호모폴리머 관련 오류에 대응하기 위해 특별히 고안되어 구현된 방법들이 있다.
호모폴리머 오류는 HomoPolish103, NanoPolish80, Pilon86 같은 일부 도구를 사용해 어셈블리를 polishing할 때 특별히 고려된다. nanopore 시퀀싱에서 호모폴리머 구간의 base calling을 개선하기 위한 방법도 개발되었고104,105, guppy나 scrappie 같은 최첨단 base-caller에도 구현되어 있다53.
시퀀싱 이전 단계에서도 이러한 오류의 영향을 줄이기 위한 조치를 취할 수 있는데, 바코드 서열에서 호모폴리머를 피하는 것106,107이나 DNA 기반 저장 시스템 개발 과정에서 호모폴리머를 회피하는 것108 등이 그 예이다.
시퀀싱 기술을 개선하는 것 역시 해결책이 될 수 있으며, 소스 단계에서 호모폴리머 오류의 수를 줄일 수 있다. 최신 ONT 화학 R.10은 호모폴리머가 풍부한 영역에서 정확도를 개선한다고 보고되었다74,109. 비생물학적 고체 상태(solid-state) nanopore 역시 호모폴리머에서 오류를 줄인다110,111.
호모폴리머 오류는 read-mapping 같은 후속 분석에 해로울 수 있다(예: 3장). 그러나 많은 경우 read를 더 최신 기술로 재시퀀싱하거나, 더 나은 base-caller로 다시 base calling할 수 없다. 호모폴리머 오류를 포함할 수 있는 read 서열만 사용 가능하다. 이러한 유형의 오류를 고려하기 위해 간단한 전처리 요령이 개발되었는데, 바로 homopolymer compression(HPC)이다.
아이디어는 매우 간단하다. 어떤 서열이든, 반복된 뉴클레오타이드 런(즉 호모폴리머)을 그 뉴클레오타이드의 단일 등장으로 치환한다. 이는 HPC를 적용하면 AAACTGGG가 ACTG로 변함을 의미한다. 모든 read와 분석할 서열에 이 간단한 전처리를 적용하면 호모폴리머 내의 모든 indel이 제거되어 일부 모호성이 해소될 수 있다(그림 1.6 참조). 호모폴리머에 담긴 정당한 정보가 제거될 수도 있지만, 오류율 감소와의 트레이드오프는 유리하다고 여겨져 왔다.
HPC는 많은 서열 생물정보학 소프트웨어 도구에 구현되어 있다. HiCanu112, MDBG113, wtdbg275, shasta114 같은 어셈블러는 더 나은 어셈블리를 위해 내부적으로 HPC를 사용하며, 완전 인간 유전체 서열을 어셈블하는 데도 HPC가 사용되었다4. HPC가 처음 출판된 사용 사례는 pyrosequencing read를 위해 개발된 CABOG 어셈블러였다115. HPC는 클러스터링116, LSC117 및 LSCPlus118를 통한 롱리드 오류 교정, minimap2119 및 winnowmap2120를 통한 정렬, 위성 탠덤 반복(satellite tandem repeats)을 위한 특화 분석 파이프라인121 등 다른 작업에도 구현되어 있다.
![Image 6: 호모폴리머 압축은 시퀀싱 오류로 인한 모호성을 해소하는 데 도움이 될 수 있다.호모폴리머 관련 시퀀싱 오류가 있는 read는 기준 유전체의 두 서로 다른 구간과 상동일 수 있으며, 각 구간에 대해 불일치가 하나씩 생길 수 있다. HPC를 적용하면 이 모호성이 적절히 반영되어 read는 한 구간에만 상동이 된다. 다만 이 그림은 호모폴리머가 해로울 수 있는 한 가지 방식만을 보여주며 다른 경우도 가능하다^[호모폴리머 indel은 반대 상황에서도 해로울 수 있다. 예를 들어, 보존된 모티프의 여러 반복에 대응해야 하는 read를 생각해보자. 호모폴리머 indel은 read를 유일하게 만들어 모호성을 인위적으로 해소하고 모티프의 특정 반복을 선호하게 만들거나, read를 완전히 잘못된 위치에 배치할 수 있다.].](https://thesis.lucblassel.com/figures/Sequence-Intro/Hpc.png)
Figure 1.6: 호모폴리머 압축은 시퀀싱 오류로 인한 모호성을 해소하는 데 도움이 될 수 있다.
호모폴리머 관련 시퀀싱 오류가 있는 read는 기준 유전체의 두 서로 다른 구간과 상동일 수 있으며, 각 구간에 대해 불일치가 하나씩 생길 수 있다. HPC를 적용하면 이 모호성이 적절히 반영되어 read는 한 구간에만 상동이 된다. 다만 이 그림은 호모폴리머가 해로울 수 있는 한 가지 방식만을 보여주며 다른 경우도 가능하다3.
이 장을 읽고 나면, 시퀀싱이 생물학적 과정, 생물체, 그리고 생명 전반에 대한 우리의 지식을 확장하는 데 근본적이라는 점에 동의하게 되기를 바란다. 그러므로 시퀀싱 분야는 여전히 매우 활발하며, 다양한 측면에서 현 기술을 개선하기 위한 새로운 기술이 개발되고 있다. Illumina는 Infinity97로 고정확도 롱리드를 약속하고, PacBio는 sequencing by synthesis에서 벗어나 자체적인 숏리드 시퀀싱 기술을 개발 중이다122,123. 또한 Ultima genomics는 기가베이스당 $1까지 낮은 비용으로 정확한 숏리드를 약속하며124, 시퀀싱을 더 저렴하고 더 많은 환경에서 이용 가능하게 만들기 위한 노력도 이어지고 있다.
이러한 기술적 개선으로 우리는 시퀀싱이 쉽고 빠른 시대에 다가가고 있으며, Tara Oceans125나 BioGenome project126 같은 대규모 프로젝트가 생물다양성을 더 잘 이해할 수 있는 문을 열고 있다. 정기적인 전장 유전체 시퀀싱은 개인 맞춤 의학의 시대를 열 수도 있다127.
이러한 발전에도 불구하고 시퀀싱 오류는 특정 분석에서 여전히 장애물이다. 이는 점점 더 널리 쓰이고 유용한 롱리드와, 유전체에서 중요한 비중을 차지하는 호모폴리머 구간에서 특히 그렇다. 어떤 방식으로든 이러한 오류를 탐지하고 제거하거나 고려하는 것은 시퀀싱 데이터에 기반한 모든 분석을 개선하고, 잘못된 서열 데이터 위에 이론이나 결론이 세워지는 일을 방지하기 위한 핵심 단계이다.
마지막으로(적어도 이 논문의 나머지 부분에서는) 계산 관점에서 생물학적 서열은 단지 문자들의 연속이며, read 집합은 단지 텍스트 파일이라는 점을 강조할 필요가 있다. 따라서 많은 분석과 데이터 처리 방법은 텍스트 알고리즘 분야에서 영감을 받거나 그로부터 직접 전이되었다.
Watson, J. D. & Crick, F. H. C. The Structure of Dna. Cold Spring Harb Symp Quant Biol18, 123–131 (1953).
Macgregor, H. C. C-Value Paradox. in Encyclopedia of Genetics (eds. Brenner, S. & Miller, J. H.) 249–250 (Academic Press, 2001). doi:10.1006/rwgn.2001.0301.
Kiefer, J. Effects of Ultraviolet Radiation on DNA. in Chromosomal Alterations: Methods, Results and Importance in Human Health (eds. Obe, G. & Vijayalaxmi) 39–53 (Springer, 2007). doi:10.1007/978-3-540-71414-9_3.
Bennett, J. W. & Klich, M. Mycotoxins. Clin Microbiol Rev16, 497–516 (2003).
Kantidze, O. L., Velichko, A. K., Luzhin, A. V. & Razin, S. V. Heat Stress-Induced DNA Damage. Acta Naturae8, 75–78 (2016).
Pollard, M. O., Gurdasani, D., Mentzer, A. J., Porter, T. & Sandhu, M. S. Long reads: Their purpose and place. Human Molecular Genetics27, R234–r241 (2018).
Smith, B. J. Protein Sequencing Protocols. (Springer Science & Business Media, 2002). doi:10.1385/1592593429.
Wang, Y., Zhao, Y., Bollas, A., Wang, Y. & Au, K. F. Nanopore sequencing technology, bioinformatics and applications. Nat Biotechnol39, 1348–1365 (2021).
Tischler, G. & Myers, E. W. Non Hybrid Long Read Consensus Using Local De Bruijn Graph Assembly. 106252 (2017) doi:10.1101/106252.
Sanderson, N. et al. Comparison of R9.4.1/Kit10 and R10/Kit12 Oxford Nanopore flowcells and chemistries in bacterial genome reconstruction. 2022.04.29.490057 (2022) doi:10.1101/2022.04.29.490057.
Booeshaghi, A. S. & Pachter, L. Pseudoalignment facilitates assignment of error-prone Ultima Genomics reads. 2022.06.04.494845 (2022) doi:10.1101/2022.06.04.494845.
Sarkozy, P., Jobbágy, Á. & Antal, P. Calling Homopolymer Stretches from Raw Nanopore Reads by Analyzing k-mer Dwell Times. in Embec&Nbc 2017 (eds. Eskola, H., Väisänen, O., Viik, J. & Hyttinen, J.) 241–244 (Springer, 2018). doi:10.1007/978-981-10-5122-7_61.
Almogy, G. et al. Cost-efficient whole genome-sequencing using novel mostly natural sequencing-by-synthesis chemistry and open fluidics platform. 2022.05.29.493900 (2022) doi:10.1101/2022.05.29.493900.