한 가족이 유전 질환의 희귀한 원인을 찾는 3년간의 과정을 담은 실화입니다. 과학적 희망, 절망, 진단의 여정이 펼쳐집니다.
나는 내 아들의 죽음의 원인을 찾았다.
3년이 걸렸다.
하지만 우리는 해냈다.
 이렇게 극적이진 않다.
이렇게 극적이진 않다.
한 가지는 명확히 해야 한다: 내 아들은 아직 살아 있다.
그럼에도, 아내 크리스티나와 나는 그의 죽음에 책임이 있다고 판정을 받았다.
우리 아들 버트랑에겐 새로운 유전 질환이 있다.
환자 0번.
그것을 찾기 위해, 듀크 대학교의 과학자 팀이 나, 아내, 그리고 아들에게 전체 엑솜 시퀀싱(단백질 생성과 관련된 유전체 일부만을 분석)을 실시했다.
우리는 아들이 같은 유전자(NGLY1, N-Glycanase 1 효소를 생산함)에 2개의 서로 다른 (지금까지 알려진 바로는 유일한) 돌연변이를 부모로부터 각각 물려받았음을 발견했다. 그 결과, 이 효소를 만들어낼 수 없다.
우리 아들은 이 효소가 결핍된 것이 확인된 유일한 인간이다.
아래는 우리가 가장 희박한 진단에 이르기까지의 여정을 기록한 것이다.
이 이야기는 오직 과학이 줄 수 있는 희망에 관한 이야기다.
업데이트: The Journal of Medical Genetics의 오픈 액세스 논문에 이 획기적인 실험의 상세 결과가 실렸다.
업데이트: 이 글을 쓴 이후, 전 세계적으로 NGLY1 결핍 사례가 2 8 15건 더 발견됐다(엑솜/유전체 시퀀싱으로 확인됨). 만약 소견이 다른 환자를 발견한다면 양상이 다양할 수 있으니 본문의 환자와 다를 수 있다. 의심되면 반드시 연락해달라. 모든 환자 정보는 비밀로 한다.
업데이트: 또 다른 부모와 함께, 진단 과정을 담은 Genetics in Medicine 논문과 8명의 NGLY1 결핍 환자의 임상 양상을 기술한 논문도 출판되었다.
업데이트: 이 글 작성 2년 후, The New Yorker에서 Seth Mnookin이 후속기를 썼다.
번역본:
심한 황달을 제외하면, 버트랑은 출생 시 정상적이었다.
두 달 동안은 정상적으로 성장했다.
세 달이 됐을 때, 발달 속도가 느려졌지만 "정상 범위 내"였다.
생후 6개월, 운동 기능이 거의 없었다.
우리는 그를 "흔들린다(jiggly)"고 표현했다.
뭔가 잘못된 것이었다.
버트랑이 처음 발달 소아과 전문의를 만난 것은 생후 8개월, 유타로 이사온 직후였다.
나는 첫 교직원 워크숍에 있었고, 끝나자 아내의 음성/문자 메시지가 잔뜩 와 있었다.
심장이 쿵쾅거렸다.
소아과 의사는 뇌 손상을 의심, 다음 주 MRI를 예약했다.
MRI는 멀쩡한 뇌를 보여줬다.
다시 소아 신경과로 의뢰됐다.
운동 장애는 확실한데, 전형적인 코레아나 운동실조는 없었다.
혈액 검사 처방.
이것이 수십 번의 피뽑기 시작이었다.
(지금은 버트랑이 좋아하는 임상병리사에게 연말 카드를 보낸다.)
혈액검사 결과는 단 하나의 이상: 알파-태아단백(AFP) 수치가 너무 높았다.
AFP 상승을 보이는 질환은 극소수.
운동 장애와 AFP 상승이 동시에 있는 건 운동실조-모세혈관확장증(A-T) 뿐.
A-T는 진행성, 치명적, 불치, 치료법 없음.
아내와 나는 충격에 빠졌다.
A-T는 상염색체 열성 유전 질환이다. 이 때문에, 우리는 수없이 물어봐야 했다:
두 분, 혹시 친척 아닙니까?
나는 오하이오 출신, 북유럽계. 아내는 다세대 푸에르토리코계.
가계도를 수세대 거슬러 올라가도 교집합 없음.
아니오.
우리는 친척이 아니다.
[참고: 생물학 공식 교육은 고등학교 2개월뿐. 오류 지적 환영.]
왜 모든 의사가 친척 여부를 묻는지, 버트랑의 진단이 얼마나 희귀한지 이해하려면 유전자와 돌연변이에 대해 알아야 한다.
유전체는 신체의 설계도다.
이 정보는 4글자(아데닌, 시토신, 티민, 구아닌)로 쓰인다: A, C, T, G.
컴퓨터의 0, 1이 인생에서 하는 역할과 같다.
컴퓨터에서 00000100이 뭔가 직접 연산을 의미하는 것처럼 DNA의 배열이 정보를 담는다.
컴퓨터에선 x86 명령셋이 있다.
삶에도 그런 명령셋, DNA 코돈 테이블이 있다. (DNA 코돈 표)
유전암호는 코돈이라는 명령어로 이뤄져 있다. 코돈은 3글자 DNA로, 아미노산 혹은 '단백질 생산 중지'를 뜻한다.
예를 들어 TTG는 '류신 추가'를 의미한다.
4글자 알파벳으로 4³=64가지 코돈이 있지만 실제 기능명령은 25가지 정도이다.
인간 유전체 내 유전자는 함수 단위다(프로그램의 함수 코드처럼). 엑손(단백질 생성에 관여)과 인트론(주석처럼 무시됨)으로 구성된다.
엑손이 아미노산 배열을 기술, 단백질로 접힌다.
유전자가 단백질로 컴파일되면, 주로 효소로서 세포 내 함수 역할을 한다.
남성과 성염색체 외에는 모든 유전자가 이중본: 하나는 엄마, 하나는 아빠. 이중 유전자는 중복성을 부여한다.
이 유전자 중복성은 돌연변이에 대한 강력한 방어막이다.
돌연변이는 유전자 코드 변경이다.
예를 들어 T를 A로, 혹은 A를 G로 바꾼다.
이런 변화는 아미노산 삽입 변화를 일으키기도 하고(TTT->TTA) 안 바뀌는 경우도 있다(TTA->TTG).
드물게, 코돈이 '정지' 명령으로 바뀌기도 한다(TGG->TGA).
이를 무의미(넌센스) 돌연변이라고 하며, 단백질 기능을 상실한다.
일부 돌연변이는 임의 수의 글자를 넣거나 뺀다. 3의 배수면 이후 코돈이 정상적으로 읽힌다(프레임 내 변이). 그렇지 않으면 유전자 이후 흐름이 엉킨다(프레임 쉬프트 변이).
대개 돌연변이가 유전자 뒤쪽에서 일어나면 덜 심각하다. (하지만, 최후단 프레임 쉬프트도 치명적일 수 있음을 배웠다.)
돌연변이 발생 시 네 가지 가능성:
A-T와 같은 질환은 무해하지만 비기능적 유전자가 만났을 때 발생한다.
돌연변이의 후손들이 만날 확률이 높은 집단/지리적 환경에서 유행한다.
둘 다 같은 변이 유전자를 받으면 열성질환 발현. (대개 먼 친척들.)
하지만, 우리는 친척이 아니다. 그럼에도, 버트랑은 상염색체 열성 유전 질환.
버트랑은 엄마를 더 닮았다.
우리가 친척 아님을 확신 있게 말한 후, 이후 몇 년간 거의 모든 의사/간호사가 아내를 따로 불러 "혹시 남편이 친부 아닌 건 아닌가요?"라고 물었다.
아니다.
A-T 진단의 충격에서 벗어나자 우리는 조사에 들어갔다.
며칠도 안 되어 버트랑이 A-T가 아님을 확신했다.
증상은 일치하지 않았다.
유전자 검사에서 A-T 음성.
10개월, 버트랑은 증상 교집합의 공집합에 들어갔다.
새 발견이 나올 때마다 공집합은 더 텅 비었다.
그 뒤 수개월간 사소한 결과들이 나왔다.
그때마다 새 가설과 (부정적인) 테스트가 반복됐다.
ALT, AST 수치 상승으로 간 기능 장애를 의심했지만, 소화기내과 정밀검진은 이상 없었다.
정신적 발달은 8개월 무렵 멈춰, 현재(4세)까지 그렇다.
버트랑의 특이한 사례는 전문가들의 관심을 불렀다.
진단에 성공한 이 없음.
15개월 무렵 결정적 소견, 소변에서 올리고당 발견. 이는 선천성 대사 이상 가족을 암시한다.
(우린 이후 새로운 카테고리를 알게 됐다: _탈_당화(glycosylation) 장애.)
예상 수명 2~3년으로 줄어듦.
울트라-레어 희귀질환 목록들... (중략)
이런 질환의 대부분은 효소 결핍이 원인.
이론적으로, 이 효소를 투여하면 진행 멈출 수 있다.
효소를 합성할 수 있더라도, 전달이 어렵다. (혈액-뇌 장벽 때문.)
대부분 경우, 인간은 효소 합성을 모른다. 결국, 수혈을 통한 골수 이식이 유일한 방법.
골수 이식은 기존 뼈속 줄기세포를 완전 제거 후, 기증자의 골수로 대체.
줄기세포가 퍼지면서, 환자는 두 유전자를 지닌 키메라가 된다.
기증자 세포가 결손 효소 생산 시 정상 기능 회복 기대.
올리고당 발견 후, 우리는 본격적으로 어떤 효소 결핍 질환인지 좁혀가기 시작했다.
Dr. Joanne Kurtzberg 교수와 상담. 위험 부담 30%의 이식에 앞서, 확실한 진단 원했다.
그래서, Dr. Vandana Shashi, Kelly Schoch 유전학자와 만남. 지금도 계속 협업 중.
듀크 신경팀이 EEG와 MRI 추가 시행. "이상한", "아마도 간질성" 뇌파 활동 확인됨.
MRI상 뇌 백질(기능적 네트워크)의 지연성 수초화, 혹은 손실. 이는 대사 이상에 의한 백질이영양증과 양상 일치.
듀크를 떠나기 전, Dr. Kurtzberg는 더는 버트랑이 이식의 효과를 보기 어려울 것이라 진솔하게 말했다. 우리는 충격을 받았다.
듀크 이후, 진단보다는 치료에 주력하기 시작했다. 이상운동의 다수가 간질임을 알게 됐다. 근간대성, 결신, 이완성 등 3가지 종류의 발작이 있었다.
곧 긴장성 간질이 추가. 항간질약 Keppra 투여.
Keppra가 일부 효과에 그치자 케토제닉 식이 시도. 뇌를 포도당 대신 케톤으로 전환시키는 고지방 식단. 발작이 거의 사라짐. 하지만, 긴장성 발작 발생으로 대안 모색.
2세 무렵, 버트랑이 울어도 눈물이 없음을 알아챔. 검색 결과 Allgrove 증후군에 도달. 유전자 검사 음성.
Dr. Stratakis가 버트랑에게 흥미를 보임. NIH 패널서 여러 질환 감별, 유전자 검사. 최종적으로 남성 Rett 증후군 의심. 그러나 추가 검사 모두 음성.
고용량 ACTH 투여가 일부 난치성 간질에 효과. 운 좋게도, 버트랑에게도 의외로 듣는다. 하지만 극심한 부작용: 비만, 탈모, 분노, 조숙.
ACTH와 케토식이는 면역 기능 저하, 중증 호흡기 감염으로 중단. 전신 부종, 움직이지 못함. 생명이 위태로웠다. 각종 항생제, 인공호흡기 치료.
ACTH와 케토식이 중단 다음 날, 병상에서 처음으로 버트랑이 웃었다. 커다란 변화였다. 아내는 눈물을 흘렸다.
회복 후, 2개월간 발작 없다. 발달도 재개. 인생 최고의 두 달간.
마이오클로닉 발작만 돌아옴. 라미탈 추가 처방.
입원 중 간 생검 실시. 간섬유화 발견. 우르소디올 시도. 조금 호전.
심전도에서 롱QT 증후군 소견. 드물지만 치명적 유전성 문제. 한동안 채널로파시(gene channelopathy)도 의심함.
모든 진단 가능성을 거의 소진, 우리는 버트랑의 변이가 de novo(새 돌연변이), 즉 우리가 아닌 버트랑에게서 최초로 생긴 것이라 추측. 그래서 둘째 임신에 대한 위험이 없을 거라 생각했으나, 오판이었다.
유타대 Dr. Lynn Jorde와 만나 3인 유전체 전체 시퀀싱 가능성 논의. 비용 문제-시간상 어려움 드러남.
듀크(Dr. Shashi와 Kelly Schoch)에서 새 기술인 엑솜만 분석하는 방법 제안. 전체 유전체의 약 2%만 분석하면 대부분의 유전질환 돌연변이 찾을 수 있다. 버트랑과 11명의 미진단 아동이 파일럿 연구 참여.
간 치료와 모니터링으로 1년만에 간 수치 정상화. 롱QT 문제 역시 선천이 아닌 약물유발임(특히 입원 중 복용한 에리트로마이신 때문) 확인.
심장과 간이 호전된 직후, 심각한 안구 감염. 눈물이 없으니 각막이 손상됨. 항생제와 인공안약, 연고로 관리. 하루 2시간마다 필요. 반나절만 소홀해도 1주일 이상 감염됨.
엑솜 실험 중, 둘째 임신. 실험 결과가 둘째에게 위험을 알려줄 수 있지만 어떠한 정보도 임신 진행 판단에 사용하지 않기로 결정.
버트랑의 돌연변이가 체세포 모자이크일 가능성 가정. 태어날 때 채취한 제대혈 줄기세포 주입. 임상 연구 참여. 돌연변이가 제대혈에 없다면 어떤 재생 효과를 기대.
수술은 불가능하다 들었으나, 확실히 확인 위해 방문. MRI상 백질 감소가 (일시적으로나마) 정지되어 보임. 줄기세포 효과일 수도 있다는 가능성.
3년 반, 둘째 빅토리아 출산 1주 전, 듀크 방향에서 연락. 다만 IRB 때문에 정보를 알려주지 못함. 우리가 X염색체 변이로 추정(빅토리아는 이 경우 영향 없음). 하지만, 실제로는 아니었다.
아내 아버지도 같은 변이. 따라서 범인 아님 확정.
건성안의 원인으로 비타민 결핍 검색. 검진 결과 간에 저장되는 비타민이 부족. 고용량 보충으로 눈물 조금 나오기 시작.
4년 반, 듀크에서 연락. 엑솜 연구가 끝났고, 답을 찾았다.
나는 8번째 엑손에서 nonsense(종결) 변이, 아내는 마지막 엑손에서 프레임쉬프트. 각각 정상 생산의 절반만 만듦. 버트랑은 둘 다 물려받아 0이 됨:
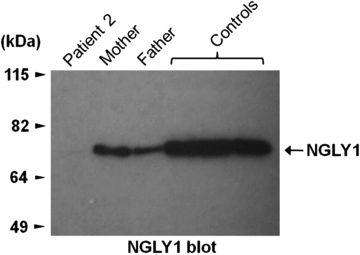
진한 띠가 N-Glycanase 1의 양. 버트랑은 "Patient 2."
그는 이 효소가 없는 첫 번째이자 유일한 인간이다:
N-Glycanase 1은 잘못 접힌 당단백질을 재활용하도록 deglycosylation 한다. 버트랑의 세포엔 잘못 접힌 glycoprotein이 쌓이는 것으로 보인다.
이 진단의 확률을 곰곰 생각해볼 만하다. 우리의 돌연변이는 모두 수천 명의 대조군에서 유일하다. 즉, 1천명당 1명, 무작위 결합 시 100만분의 1. 둘 다 유전자 증상을 가질 확률은 4백만분의 1. 앞으로 더 비슷한 NGLY1 변이나 환자가 발견될 때까지, 이 확률은 계속 낮아질 것. 빅토리아가 둘 다 받을 확률은 4분의 1이었으나, 둘 다 없다.

아직 한 살도 안 된 빅토리아가 버트랑을 휠체어에 태워 학교 버스정류장에 데려다준다.
아내의 아버지 Dr. Manuel Casanova와 친구 Dr. Karen Ho와 함께 N-Glycanase 1 결핍의 생물학적 의미 분석 시작. Dr. Ho는 이 결핍이 소포체 스트레스를 일으킬 수 있다고 가정, 항산화제의 효과 검토. 아내 아버지는 내 nonsense 변이를 읽어내는 신약 gentamicin 검토.
며칠 후 아내 자체 연구로, N-Glycanase 1의 한 변형이 이미 합성이 가능하다는 사실(Genzyme이 특허 보유)에 도달. 실험실 용도로 이미 쓰이고 있음. 주문하면 $244.
불행히도, 그냥 구입해서 주사할 순 없다. FDA 승인, Genzyme 협조, 안전성-생체 이용률 등 확인 및 준비 필요. 과정은 험난하겠으나 아들의 생명이 달려 있다. 우리는 계속할 것이다.
올리고당 발견을 알리던 블로그 글에서 우리는 버트랑에게 약속했다:
그에게 가능한 모든 것을 시도하겠다. 만약 그게 안 된다면, 불가능 같은 일도 도전하겠다.
진정한 '불가능'을 시도한다는 건 무엇일까?
박사과정 만화 안내서에서 나는 인간 지식의 경계에 틈을 내는 것에 대해 이야기했다. 이 글은 그 중 하나, 현대 과학의 본질적인 과정을 담고 있다.
과학은 알려지지 않은 것을 알려진 것으로 바꾸는 체계적인 과정이다. 즉, 불가능을 가능으로 바꾼다.
그 안내서 이후, 나는 그 변화를 강조하는 에필로그를 추가했다:

경계에는 새로운 틈이 뚫렸다.
우린 거의 왔다. 해야 할 일은 계속 밀어붙이는 것뿐.
업데이트: 이 글 작성 2년 후 The New Yorker에서 Seth Mnookin이 후속기를 썼다.